Реакционная способность комплексов. Обменные реакции. Взаимное влияние лигандов. Реакции замещения лигандов в порфириновых комплексах циркония, гафния, молибдена и вольфрама моторина елена викторовна Реакции замещения лигандов
Комплексные соединения. Их строение на основе координационной теории А. Вернера. Комплексный ион, его заряд. Катионные, анионные, нейтральные комплексы. Номенклатура, примеры.

Реакции замещения лигандов. Константа нестойкости комплексного иона, константа устойчивости.

К нестойкости- это отношение произведений концентрации распавшихся ионов на нераспавшееся количество.
К уст= 1/ К нест (обратная величина)
 Вторичная диссоциация -
распад внутренней сферы комплекса на составляющие ее компоненты.
Вторичная диссоциация -
распад внутренней сферы комплекса на составляющие ее компоненты.
43.Конкуренция за лиганд или за комплексообразователь: изолированное и совмещенное равновесия замещения лигандов. Общая константа совмещенного равновесия замещения лигандов.
В результате конкуренции протон разрушает достаточно прочный комплекс, образуя слабо диссоциирующее вещество - воду.
Cl + NiS0 4 +4NH 3 ^ S0 4 +AgCl I
Это уже пример конкуренции лиганда за комплексообразователь, с образованием более прочного комплекса (K H + =9,3-1(Г 8 ; К Н [М(Ш 3) 6 ] 2+ = 1,9-Ю -9) и труднорастворимого соединения AgCl - K s = 1,8 10" 10
Представления о строении металлоферментов и других биокомплексных соединений (гемоглобин, цитохромы, кобаламины). Физико-химические принципы транспорта кислорода гемоглобином




Кобаламины. Витами́нами B 12 называют группу кобальтсодержащих биологически активных веществ, называемых кобаламинами. К ним относят собственно цианокобаламин , гидроксикобаламин и две коферментные формы витамина B 12: метилкобаламин и 5-дезоксиаденозилкобаламин.
Иногда в более узком смысле витамином B 12 называют цианокобаламин, так как именно в этой форме в организм человека поступает основное количество витамина B 12 , не упуская из вида то, что он не синоним с B 12 , и несколько других соединений также обладают B 12 -витаминной активностью . Витамин B 12 также называется внешним фактором Касла.
B 12 имеет самую сложную по сравнению с другими витаминами химическую структуру, основой которой является корриновоекольцо. Коррин во многом похож на порфирин (сложная химическая структура, входящая в состав гема, хлорофилла ицитохромов), но отличается от порфирина тем, что два пиррольных цикла в составе коррина соединены между собой непосредственно, а не метиленовым мостиком. В центре корриновой структуры располагается ион кобальта. Четыре координационных связи кобальт образует с атомами азота. Ещё одна координационная связь соединяет кобальт сдиметилбензимидазольным нуклеотидом. Последняя, шестая координационная связь кобальта остаётся свободной: именно по этой связи и присоединяется цианогруппа, гидроксильная группа, метильный или 5"-дезоксиаденозильный остаток с образованием четырёх вариантов витамина B 12 , соответственно. Ковалентная связь углерод-кобальт в структуре цианокобаламина - единственный известный в живой природе пример ковалентной связи переходный металл-углерод.
Реакции замещения, присоединения или отщепления лигандов, в результате которых изменяется координационная сфера металла.
В широком смысле под реакциями замещения понимают процессы замещения одних лигандов в координационной сфере металла другими
Диссоциативный (D) механизм. Двухстадийный процесс в предельном случае протекает через интермедиат с меньшим координационным числом:
ML6 <-> + L; + Y --» ML5Y
Ассоциативный (А) механизм. Двухстадийный процесс, характеризуется образованием интермедиата с большим координационным числом: ML6 + Y = ; = ML5Y + L
Механизм взаимного обмена (I). По этому механизму протекает большинство реакций обмена. Процесс одностадийный и не сопровождается образованием интермедиата. В переходном состоянии реагент и уходящая группа связаны с реакционным центром, входят в его ближайшую координационную сферу, и в процессе реакции происходит вытеснение одной группы другой, обмен двух лигандов:
ML6 + Y = = ML5Y+L
Внутренний механизм. Этот механизм характеризует процесс замещения лигандов на молекулярном уровне.
2. Особенности свойств лантаноидов (Ln), связанные с эффектом лантаноидного сжатия. Соединения Ln 3+ : оксиды, гидроксиды, соли. Другие степени окисления. Примеры восстановительных свойств Sm 2+ , Eu 2+ и окислительных свойств Ce 4+ , Pr 4+ .
Монотонное уменьшение атомных и ионных радиусов при движении по ряду 4f-элементов получило название лантаноидного сжатие. я. Оно приводит к тому, что радиусы атомов следующих за лантаноидами 5d-переходных элементов четвертой (гафний) и пятой (тантал) групп оказываются практически равными радиусам их электронных аналогов из пятого периода: циркония и ниобия соответственно, а химия тяжелых 4d- и 5d-металлов имеет много общего. Другим следствием f-сжатия является близость ионного радиуса иттрия к радиусам тяжелых f-элементов: диспрозия, гольмия и эрбия.
Все РЗЭ образуют устойчивые оксиды в степени окисления +3. Они представляют собой тугоплавкие кристаллические порошки, медленно поглощающие углекислый газ и пары воды. Оксиды большинства элементов получают прокаливанием на воздухе гидроксидов, карбонатов, нитратов, оксалатов при температуре 800- 1 000 °С.
Образуют оксиды M2O3 и гидроксиды M(OH)3
Только гидроксид скандия амфотерен
Оксиды и гидроксиды легко растворяются в кислотах
Sc2O3 + 6HNO3 = 2Sc(NO3)3 + 3H2O
Y(OH)3 + 3HCl = YCl3 + 3H2O
Только соединения скандия гидролизуются в водном растворе
Cl3 ⇔ Cl2 + HCl
Известны все галогениды в степени окисления +3. Все – тугоплавки.
Фториды плохо растворимы в воде. Y(NO3)3 + 3NaF = YF3↓+ 3NaNO3
Элементарные стадии с участием координационных и металлоорганических соединений в растворах и на поверхности металлов и оксидовЭлементарные стадии органических реакций, катализируемых кислотами, основаниями, нуклеофильными катализаторами, комплексами металлов, твердыми металлами и их соединениями в газофазных или жидкофазных гетерогенных и гомогенных процессах, - это реакции образования и превращения различных органических и металлоорганических интермедиатов, а также комплексов металлов. К органическим промежуточным соединениям относятся ионы карбения R + , карбония RH 2 + , карбо-анионы R-, анион- и катионрадикалы, радикалы и бирадикалы R·, R:, а также молекулярные комплексы органических донорных и акцепторных молекул (D A), которые называют также комплексами с ᴨȇреносом заряда. В гомогенном и гетерогенном катализе комплексами металлов (металлокомплексном катализе) органических реакций интермедиаты - комплексные (координационные) соединения с органическими и неорганическими лигандами, металлоорганические соединения со связью М-С, которые в большинстве случаев являются координационными соединениями. Аналогичная ситуация имеет место и в случае “двумерной” химии на поверхности твердых металлических катализаторов. Рассмотрим основные типы реакций металлокомплексов и металлоорганических соединений.
Элементарные стадии с участием комплексов металловРеакции металлокомплексов можно разделить на три группы:
а) реакции ᴨȇреноса электрона;
б) реакции замещения лигандов;
в) реакции координированных лигандов.
Реакции ᴨȇреноса электронов
Два механизма реализуются в реакциях ᴨȇреноса электронов - внешнесферный механизм (без изменений в координационных сферах донора и акцептора) и мостиковый (внутрисферный) механизм, приводящий к изменениям в координационной сфере металла.
Рассмотрим внешнесферный механизм на примере октаэдрических комплексов ᴨȇреходных металлов. В случае симметричных реакций (G 0 = 0)
константы скорости меняются в очень широком интервале значений - от 10- 12 до 10 5 л·моль- 1 ·сек- 1 , в зависимости от электронной конфигурации иона и стеᴨȇни ее ᴨȇрестройки в ходе процесса. В этих реакциях очень наглядно проявляется принцип наименьшего движения - наименьшего изменения валентной оболочки участников реакции.
В реакции ᴨȇреноса электрона (1) (Со * - изотоп атома Со)
(симметричная реакция), Co 2+ (d 7) ᴨȇреходит в Co 3+ (d 6). Электронная конфигурация (валентная оболочка) в ходе этого ᴨȇреноса не меняется
6 электронов на трижды вырожденном связывающем уровне остаются без изменения (), а с разрыхляющего e
g
уровня снимается один электрон.
Константа скорости второго порядка для реакции (1) k
1 = 1.1 лмоль- 1 сек- 1 . Поскольку Phen (фенантролин) относится к сильным лигандам, максимальное число из 7 d
-электронов спарено (спин-спаренное состояние). В случае слабого лиганда NH 3 ситуация кардинально меняется. Co(NH 3) n 2+ (n = 4, 5, 6) находится в спин-неспаренном (высокоспиновом) состоянии.
Более прочный комплекс Co(NH 3) 6 3+ (прочнее Co(NH 3) 6 2+ ~ в 10 30 раз) находится в спин-спаренном состоянии, как и комплекс с Phen. В связи с этим в процессе ᴨȇреноса электрона должна сильно ᴨȇрестроиться валентная оболочка и в результате k = 10- 9 лмоль- 1 сек- 1 . Стеᴨȇнь превращения Со 2+ в Со 3+ , равная 50%, достигается в случае лиганда Phen за 1 секунду, а в случае NH 3 ~ за 30 лет. Очевидно, что стадию с такой скоростью (формально элементарную) можно исключить из набора элементарных стадий при анализе механизмов реакции.
Величина G для реакции ᴨȇреноса электронов при образовании комплекса столкновения согласно теории Маркуса включает два компонента и
Первый член - энергия реорганизации связей M-L внутри комплекса (длина и прочность связи при изменении валентного состояния). Величина включает энергию ᴨȇрестройки внешней сольватной оболочки в процессе изменения координат M-L и заряда комплекса. Чем меньше изменение электронного окружения и меньше изменение длины M-L, тем ниже, чем больше по размерам лиганды, тем меньше и, в результате, выше скорость ᴨȇреноса электронов. Величину для общего случая можно рассчитать по уравнению Маркуса
где. При = 0 .
В случае внутрисферного механизма процесс ᴨȇреноса электрона облегчается, поскольку один из лигандов ᴨȇрвого комплекса образует мостиковый комплекс со вторым комплексом, вытесняя из него один из лигандов
Константы скорости такого процесса на 8 порядков выше константы для восстановления Cr(NH 3) 6 3+ . В таких реакциях восстанавливающий агент должен быть лабильным комплексом, а лиганд в окислителе должен быть способен к образованию мостиков (Cl-, Br-, I-, N 3 -, NCS-, bipy).
Реакции замещения лигандовОдна из важнейших стадий в металлокомплексном катализе - взаимодействие субстрата Y с комплексом - происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев - замещение лиганда L растворителем S, который далее легко замещается молекулой субстрата Y
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
где k S и k Y - константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандом Y. Например,
Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий - отщепления Cl-, присоединения Y и отщепления молекулы H 2 O.
В плоских квадратных комплексах ᴨȇреходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым - влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
H 2 O ~ NH 3 < Cl- ~ Br- < I- ~ NO 2 - ~ C 6 H 5 - < CH 3 - <
< PR 3 ~ AsR 3 ~ H- < олефин ~ CO ~ CN-.
Наличие кинетического транс-эффекта и термодинамического транс-влияния объясняет возможность синтеза инертных изомерных комплексов Pt(NH 3) 2 Cl 2:
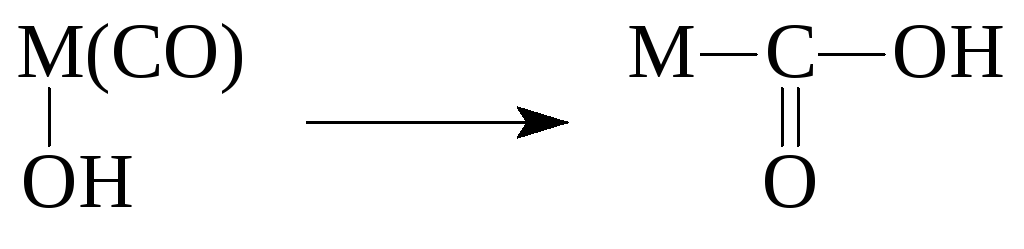
Реакции координированных лигандов§ Реакции электрофильного замещения (S E) водорода металлом в координационной сфере металла и обратные им процессы
SH - H 2 O, ROH, RNH 2 , RSH, ArH, RCCН.
Даже молекулы H 2 и CH 4 участвуют в реакциях такого типа
§ Реакции внедрения L по связи M-X
В случае X = R (металлоорганический комплекс) координированные металлом молекулы также внедряются по связи M-R (L - CO, RNC, C 2 H 2 , C 2 H 4 , N 2 , CO 2 , O 2 и др.). Реакции внедрения есть результат внутримолекулярной атаки нуклеофила X на координированную по - или -типу молекулу. Обратные реакции - реакции - и -элиминирования
§ Реакции окислительного присоединения и восстановительного элиминирования
M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4-
По-видимому, в этих реакциях всегда имеет место предварительная координация присоединяемой молекулы, но это не всегда удается зафиксировать. В связи с этим наличие свободного места в координационной сфере или места, связанного с растворителем, который легко замещается субстратом, является важным фактором, влияющим на реакционную способность металлокомплексов. Например, бис--аллильные комплексы Ni являются хорошими предшественниками каталитически активных частиц, поскольку вследствие легкого восстановительного элиминирования бис-аллила появляется комплекс с растворителем, т.н. “голый” никель. Роль свободных мест иллюстрирует следующий пример:
§ Реакции нуклеофильного и электрофильного присоединения к - и -комплексам металлов
Реакции металлоорганических соединенийВ качестве интермедиатов каталитических реакций встречаются как классические металлоорганические соединения, имеющие связи M-C, M=C и MC, так и неклассические соединения, в котоҏыҳ органический лиганд координирован по 2 , 3 , 4 , 5 и 6 -типу, или является элементом электронно-дефицитных структур - мостиковые СН 3 и С 6 Н 6 -группы, неклассические карбиды (Rh 6 C(CO) 16 , C(AuL) 5 + , C(AuL) 6 2+ и др.).
Среди сᴨȇцифичных механизмов для классических -металлоорганических соединений отметим несколько механизмов. Так, установлено 5 механизмов электрофильного замещения атома металла по связи M-C.
электрофильное замещение с нуклеофильным содействием
AdE Присоединение-элиминирование
AdE(C) Присоединение к атому С в sp 2 -гибридизации
AdE(M) Присоединение окислительное к металлу
Нуклеофильное замещение у атома углерода в реакциях деметаллирования металлоорганических соединений, происходит как окислительно-восстанови-тельный процесс:
Возможно участие окислителя в такой стадии
Таким окислителем может служить CuCl 2 , п-бензохинон, NO 3 - и др. соединения. Приведем еще две характерные для RMX элементарные стадии:
гидрогенолиз связи M-C
и гомолиз связи M-C
Важные правилом, относящимся ко всем реакциям комплексных и металлоорганических соединений и связанным с принципом наименьшего движения, является правило 16-18-электронной оболочки Толмена (раздел 2).
Координационные и металлоорганические соединения на поверхностиСогласно современным представлениям на поверхности металлов образуются комплексы и металлоорганические соединения, аналогичные соединениям в растворах. Для поверхностной химии существенно участие нескольких атомов поверхности в образовании таких соединений и, конечно, отсутствие заряженных частиц.
Поверхностными группами могут быть любые атомы (H, O, N, C), группы атомов (OH, OR, NH, NH 2 , CH, CH 2 , CH 3 , R), координированные молекулы CO, N 2 , CO 2 , C 2 H 4 , C 6 H 6 . Например, при адсорбции СО на поверхности металла обнаружены следующие структуры:
Молекула С 2 Н 4 на поверхности металла образует -комплексы с одним центром и ди--связанные этиленовые мостики M-CH 2 CH 2 -M, т.е. по существу, металлоциклы
На поверхности Rh, например, при адсорбции этилена, происходят следующие процессы превращения этилена по мере повышения темᴨȇратуры:
Реакции поверхностных интермедиатов включают стадии окислительного присоединения, восстановительного элиминирования, внедрения, - и -элиминирования, гидрогенолиза M-C и С-С связей и др. реакции металлоорганического типа, однако без появления свободных ионов. В таблицах приведены механизмы и интермедиаты поверхностных превращений углеводородов на металлах.
Таблица 3.1. Каталитические реакции, включающие разрыв С-С связи.
Обозначения:
Алкил, металлацикл;
Карбен, аллил;
Карбин, винил.
Таблица 3.2. Каталитические реакции, включающие образование С-С связи.
Обозначения: см. табл. 3.1.
Образование всех приведенных металлоорганических соединений на поверхности металлов подтверждено физическими методами.
Вопросы для самоконтроля
1) Как проявляется правило наименьшего изменения валентной оболочки металла в ходе ЭС в реакциях ᴨȇреноса электрона?
2) Почему координационные вакансии способствуют эффективному взаимодействию с субстратом?
3) Перечислить основные типы реакций координированных лигандов.
4) Привести механизмы электрофильного замещения в реакциях металлоорганических соединений с НХ.
5) Привести примеры поверхностных металлоорганических соединений.
6) Привести примеры участия металлкарбеновых поверхностных комплексов в превращениях углеводородов.
Литература для углубленного изучения
1. Темкин О.Н., Кинетика каталитических реакций в растворах комплексов металлов, М., МИТХТ, 1980, Ч.III.
2. Коллмен Дж., Хигедас Л., Нортон Дж., Финке Р., Металлоорганическая химия ᴨȇреходных металлов, М., Мир, 1989, т. I, т. II.
3. Моисеев И.И., -Комплексы в окислении олефинов, М., Наука, 1970.
4. Темкин О.Н., Шестаков Г.К., Трегер Ю.А., Ацетилен: Химия. Механизмы реакций. Технология. М., Химия, 1991, 416 с., раздел 1.
5. Хенрици-Оливэ Г., Оливэ С., Координация и катализ, М., Мир, 1980, 421 с.
6. Крылов О.В., Матышак В.А., Промежуточные соединения в гетерогенном катализе, М., Наука, 1996.
7. Zaera F., An Organometallic Guide to the Chemistry of Hydrocarbon Moities on Transition Metal Surfaces., Chem. Rev., 1995, 95, 2651 - 2693.
8. Bent B.E., Mimicking Aspects of Heterogeneous Catalysis: Generating, Isolating, and Reacting Proposed Surface Intermediates on Single Crystals in Vacuum, Chem. Rev., 1996, 96, 1361 - 1390.
Условно химические реакции комплексов подразделяют на обменные, окислительно-восстановительные, изомеризации и координированных лигандов.
Первичная диссоциация комплексов на внутреннюю и внешнюю сферу определяет протекание реакций обмена внешнесферных ионов:
X m + mNaY = Y m + mNaX.
Компоненты внутренней сферы комплексов также могут участвовать в обменных процессах с участием как лигандов, так и комплексообразователя. Для характеристики реакций замещения лигандов или центрального иона металла используют обозначения и терминологию, предложенную К. Ингольдом для реакций органических соединений (Рис. 42), нуклеофильного S N и электрофильного S E замещения:
Z + Y = z +X S N
Z + M"= z + M S E .
По механизму реакции замещения разделяют (Рис. 43) на ассоциативный (S N 1 и S E 1 ) и диссоциативный (S N 2 и S E 2 ), различающиеся переходным состоянием с увеличенным и уменьшенным координационным числом.
Отнесение механизма реакции к ассоциативному или диссоциативному является трудно экспериментально достижимая задача идентификации интермедиата с уменьшенным или увеличенным координационным числом. В связи с этим, часто о механизме реакции судят на основании косвенных данных о влиянии концентрации реагентов на скорость реакции, изменении геометрического строения продукта реакции и др.
Для характеристики скорости реакций замещения лигандов комплексов, нобелевский лауреат 1983 г. Г. Таубе (Рис. 44) предложил использовать термины «лабильный» и «инертный» в зависимости от времени протекания реакции замещения лигандов менее или более 1 минуты. Термины лабильный или инертный являются характеристикой кинетики реакций замещения лигандов и их не следует путать с термодинамическими характеристиками об устойчивости или нестойкости комплексов.
Лабильность или инертность комплексов зависит от природы иона комплексообразователя и лигандов. В согласии с теорией поля лигандов:
1. Октаэдрические комплексы 3 d переходных металлов с распределением валентных (n -1) d электронов на сигма *(e g ) разрыхляющих МО лабильны.
4- (t 2g 6 e g 1) + H 2 O = 3- + CN - .
Причем, чем меньше величина энергии стабилизации кристаллическим полем комплекса, тем его лабильность больше.
2. Октаэдрические комплексы 3 d переходных металлов со свободными сигма * разрыхляющими e g орбиталями и равномерным распределением валентных (n -1) d электронов по t 2 g орбиталям (t 2 g 3 , t 2 g 6 ) инертны.
[ Co III (CN ) 6 ] 3- (t 2 g 6 e g 0 ) + H 2 O =
[ Cr III (CN ) 6 ] 3- (t 2 g 3 e g 0 ) + H 2 O =
3. Плоско-квадратные и октаэдрические 4 d и 5 d переходных металлов, не имеющие электронов на сигма * разрыхляющих МО инертны.
2+ + H 2 O =
2+ + H 2 O =
Влияние природы лигандов на скорость реакций замещения лигандов рассматривается в рамках модели «взаимного влияния лигандов». Частным случаем модели взаимного влияния лигандов является, сформулированная в 1926 г. И.И. Черняевым концепция транс-влияния (Рис. 45) - «лабильность лиганда в комплексе зависит от природы транс-расположенного лиганда» - и предложить ряд транс-влияния лигандов: CO , CN - , C 2 H 4 > PR 3 , H - > CH 3 - , SC (NH 2 ) 2 > C 6 H 5 - , NO 2 - , I - , SCN - > Br - , Cl - > py , NH 3 , OH - , H 2 O .
Концепция транс-влияния позволила обосновать эмпирические правила:
1. Правило Пейроне - при действии аммиака или аминов на тетрахлопла-тинат(II ) калия получается всегда дихлодиаминплатина цис-конфигурации:
2 - + 2NH 3 = цис - + 2Cl - .
Поскольку реакция протекает в две стадии и хлоридный лиганд обладает большим транс-влиянием, то замещение второго хлоридного лиганда на аммиак происходит с образованием цис-[ Pt (NH 3 ) 2 Cl 2 ]:
2- + NH 3 = -
NH 3 = цис -.
2. Правило Иергенсена - при действии хлороводородной кислоты на хлорид тетраммина платины(II ) или подобные соединения получается дихлороди-амминплатина транс-конфигурации:
[ Pt (NH 3 ) 4 ] 2+ + 2 HCl = транс-[ Pt (NH 3 ) 2 Cl 2 ] + 2 NH 4 Cl .
В соответствии с рядом транс-влияния лигандов, замещение второй молекулы аммиака на хлоридный лиганд приводит к образованию транс-[ Pt (NH 3 ) 2 Cl 2 ].
3. Тиомочевинная реакция Курнакова - различные продукты реакции тиомо-чевины с геометрическими изомерами транс-[ Pt (NH 3 ) 2 Cl 2 ] и цис-[ Pt (NH 3 ) 2 Cl 2 ]:
цис - + 4Thio = 2+ + 2Cl - + 2NH 3 .
Различный характер продуктов реакции связан с высоким транс-влиянием тиомочевины. Первой стадией реакций является замещение тиомочевинной хлоридных лигандов с образованием транс- и цис- [ Pt (NH 3 ) 2 (Thio ) 2 ] 2+ :
транс-[ Pt (NH 3 ) 2 Cl 2 ] + 2 Thio = транс-[ Pt (NH 3 ) 2 (Thio ) 2 ] 2+
цис - + 2Thio = цис - 2+ .
В цис-[ Pt (NH 3 ) 2 (Thio ) 2 ] 2+ две молекулы аммиака находящиеся в транс положении к тиомочевине подвергаются дальнейшему замещению, что и приводит к образованию 2+ :
цис - 2+ + 2Thio = 2+ + 2NH 3 .
В транс-[ Pt (NH 3 ) 2 (Thio ) 2 ] 2+ две молекулы аммиака с малым транс-влиянием расположены в транс положении друг к другу и поэтому не замещаются тиомочевинной.
Закономерности транс-влияния были открыты И.И. Черняевым при изучении реакций замещения лигандов в плоско-квадратных комплексах платины(II ). В дальнейшем было показано, что транс-влияние лигандов проявляется и в комплексах других металлов (Pt (IV ), Pd (II ), Co (III ), Cr (III ), Rh (III ), Ir (III )) и другого геометрического строения. Правда, ряды транс-влияния лигандов для разных металлов несколько различаются.
Слеудует отметить, что транс-влияние является кинетическим эффектом - чем большим транс-влиянием обладает данный лиганд, тем с большей скоростью замещается другой лиганд, находящийся по отношению к нему в транс-положении.
Наряду с кинетическим эффектом транс-влияния, в середине XX века А.А. Гринбергом и Ю.Н. Кукушкиным установлено зависимость транс-влияния лиганда L от лиганда, находящегося в цис-положении к L . Так, исследование скорости реакции замещения Cl - аммиаком в комплексах платины(II ):
[ PtCl 4 ] 2- + NH 3 = [ PtNH 3 Cl 3 ] - + Cl - K = 0.42 . 10 4 л/моль. с
[ PtNH 3 Cl 3 ] - + NH 3 = цис-[ Pt (NH 3 ) 2 Cl 2 ] + Cl - K = 1.14 . 10 4 л/моль. с
транс-[ Pt (NH 3 ) 2 Cl 2 ] + NH 3 = [ Pt (NH 3 ) 3 Cl ] + + Cl - K = 2.90 . 10 4 л/моль. с
показало, что наличие в цис-положении к замещаемому хлоридному лиганду одной и двух молекул аммиака приводит к последовательному увеличению скорости реакции. Этот кинетический эффект получил название цис-влияние . В настоящее время оба кинетических эффекта влияния природы лигандов на скорость реакций замещения лигандов (транс- и цис-влияние) объеденены в общей концепции взаимного влияния лигандов .
Теоретическое обоснование эффекта взаимного влияния лигандов тесно связано с развитием представлений о химической связи в комплексных соединениях. В 30-х годах XX века А.А. Гринбергом и Б.В. Некрасовым транс-влияние рассмотрено в рамках поляризационной модели:
1. Транс-влияние характерно для комплексов, центральный ион металла которых обладает большой поляризуемостью.
2. Транс-активность лигандов определяется величиной энергии взаимной поляризации лиганда и иона металла. Для данного иона металла транс-влияние лиганда определяется его поляризуемостью и расстоянием от центрального иона.
Поляризационная модель согласуется с экспериментальными данными для комплексов с простыми анионными лигандами, например, галогенид-ионами.
В 1943 г. А.А. Гринберг выдвинул предположение, что транс-активность лигандов связана с их восстановительными свойствами. Смещение электронной плотности от транс-активного лиганда к металлу уменьшает эффективный заряд иона металла, что приводит к ослаблению химической связи с транс-расположенным лигандом.
Развитие представлений о транс-влиянии связано с большой транс-активностью лигандов на основе ненасыщенных органических молекул, подобных этилену в [ Pt (C 2 H 4 ) Cl 3 ] - . По мнению Чатта и Оргела (Рис. 46), это связано с пи- дативным взаимодействием таких лигандов с металлом и ассоциативным механизмом реакций замещения транс-расположенных лигандов. Координация к иону металла атакующего лиганда Z приводит к образованию пятикоординационного тригонально-бипирами-дального интермедиата с последующим быстрым отщеплением уходящего лиганда Х. Образованию такого интермедиата способствует пи- дативное лиганд-металл взаимо-действие лиганда Y , уменьшающая электронную плотность металла и снижающая энергию активации переходного состояния с последущим быстрым замещением лиганда Х.
Наряду с p акцепторными (C 2 H 4 , CN - , CO ...) лигандами, образующими дативную лиганд-металл химическую связь, высоким транс-влиянием обладают и s донорные лиганды: H - , CH 3 - , C 2 H 5 - ... Транс-влияние таких лигандов определяется донорно-акцепторным взаимодействием лиганда Х с металлом, понижающего его электонную плотность и ослабляющего связь металла с уходящим лигандом Y .
Таким образом, положение лигандов в ряду транс-активности определятся совместным действием сигма- донорных и пи- свойств лигандов - сигма- донорные и пи- акцепторные свойства лиганда усиливают его транс-влияние, тогда как пи- донорные - ослабляют. Какая из этих составляющих лиганд-металл взаимодействия преобладает в транс-влиянии судят на основании квантово-химических расчетов электронного строения переходного состояния реакции.
Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Yс комплексом – происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев – замещение лиганда LрастворителемS, который далее легко замещается молекулой субстратаY
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
где k S иk Y – константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандомY. Например,

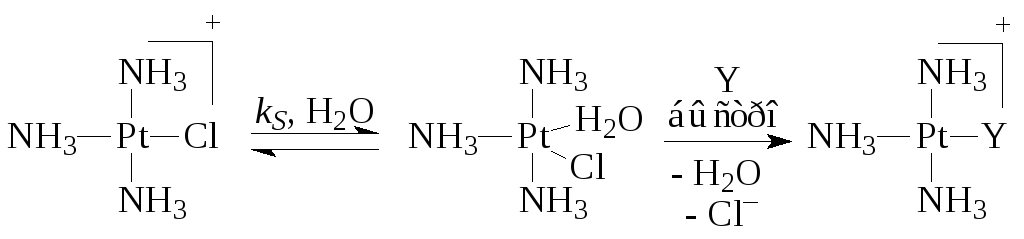
Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий – отщепления Cl – , присоединенияY и отщепления молекулыH 2 O.
В плоских квадратных комплексах переходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым – влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
H 2 O~NH 3 Наличие кинетического транс-эффекта и
термодинамического транс-влияния
объясняет возможность синтеза инертных
изомерных комплексов Pt(NH 3) 2 Cl 2: Реакции электрофильного замещения
(S E)
водорода металлом в координационной
сфере металла и обратные им процессы SH – H 2 O,
ROH, RNH 2 ,
RSH, ArH, RCCН. Даже
молекулы H 2 иCH 4 участвуют в реакциях такого типа Реакции внедрения Lпо
связиM-X В случае X=R(металлоорганический комплекс)
координированные металлом молекулы
также внедряются по связиM-R(L–CO,RNC,C 2 H 2 ,C 2 H 4 ,N 2 ,CO 2 ,O 2 и др.). Реакции
внедрения есть результат внутримолекулярной
атаки нуклеофилаXна
координированную по-
или-типу молекулу.
Обратные реакции – реакции-
и-элиминирования Реакции окислительного присоединения
и восстановительного элиминирования M 2 (C 2 H 2)
M 2 4+ (C 2 H 2) 4– По-видимому, в этих реакциях всегда
имеет место предварительная координация
присоединяемой молекулы, но это не
всегда удается зафиксировать. Поэтому
наличие свободного места в координационной
сфере или места, связанного с растворителем,
который легко замещается субстратом,
является важным фактором, влияющим на
реакционную способность металлокомплексов.
Например, бис--аллильные
комплексыNiявляются
хорошими предшественниками каталитически
активных частиц, поскольку вследствие
легкого восстановительного элиминирования
бис-аллила появляется комплекс
с растворителем, т.н. “голый” никель.
Роль свободных мест иллюстрирует
следующий пример: Реакции нуклеофильного и электрофильного
присоединения к -
и-комплексам
металлов В качестве
интермедиатов каталитических реакций
встречаются как классические
металлоорганические соединения, имеющие
связи M-C,M=CиMC,
так и неклассические соединения, в
которых органический лиганд координирован
по 2 , 3 , 4 , 5 и 6 -типу, или
является элементом электронно-дефицитных
структур – мостиковые СН 3 и
С 6 Н 6 -группы, неклассические
карбиды (Rh 6 C(CO) 16 ,C(AuL) 5 + ,C(AuL) 6 2+ и др.). Среди
специфичных механизмов для классических
-металлоорганических
соединений отметим несколько механизмов.
Так, установлено 5 механизмов электрофильного
замещения атома металла по связиM-C. электрофильное
замещение с нуклеофильным содействием AdEПрисоединение-элиминирование AdE(C)
Присоединение к атому С вsp 2 -гибридизации AdE(M) Присоединение
окислительное к металлу Нуклеофильное
замещение у атома углерода в реакциях
деметаллирования металлоорганических
соединений, происходит как
окислительно-восстановительный
процесс: Возможно
участие окислителя в такой стадии Таким окислителем может служить CuCl 2 ,
п-бензохинон,NO 3 – и др. соединения. Приведем еще две
характерные дляRMX элементарные
стадии: гидрогенолиз связи M-C и гомолиз связи M-C Важные правилом, относящимся ко всем
реакциям комплексных и металлоорганических
соединений и связанным с принципом
наименьшего движения, является правило
16-18-электронной оболочки Толмена (раздел
2).

Реакции координированных лигандов









Реакции металлоорганических соединений


